

The clinical development of oral polypeptide drugs is subject to some of their unfavorable physicochemical properties, such as molecular size, charge, stability, which hinder their passage through biological barriers such as the intestinal mucosa. Moreover, oral peptides face challenges with enzymatic degradation and stability is greatly affected. Many structural features of natural polypeptides, such as the N-terminal amino and C-terminal carboxyl, side-chain carboxyl, amino and hydroxyl groups, make them recognized by a variety of proteases in the organism and are degraded before reaching the system. They can be described as "die before starting".
In response to these inherent problems against the oral polypeptide formulation, prodrug (prodrug) strategies have been successfully used to transiently alter the physicochemical properties of drugs and improve their metabolic instability. Precursor drug, also known as prodrug and prodrug, refers to the drug agent molecule that becomes active drug ingredients after chemical reaction or enzymatic transformation in the organism. It itself belongs to a pharmacological inert substance. That is, the precursor drug itself has no biological activity or a very low activity, and becomes an active substance after metabolism in the body. The purpose of this strategy is to increase drug bioavailability, enhance targeting, and reduce drug toxicity and side effects.
Desmopressin desmopressin is a synthetic analogue of the vasopressin hormone vasopressin vasopressin, which is mainly used for the treatment of urological disorders such as nocturia and diabetes insipidus. The ester precursors of desmopressin are produced by phenolic hydroxylipidation of the tyrosine side chains. These derivatives were quantitatively converted to desmopressin by enzymatic hydrolysis in human plasma (Figure 1).[1]Its new desmopressin precursor drug transport across Caco-2 cell monolayers was significantly more efficient than desmopressin.
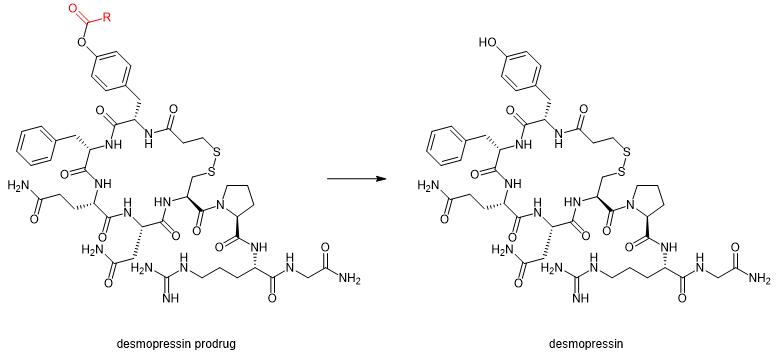
Figure 1. Desmopressin precursor drug
Biorereversible looping of polypeptides is one of the most promising new approaches for drug development for peptide precursors. Cyclization of the peptide backbone reduces the two polar groups of the peptide molecule —— N-terminal amino group and C-terminal carboxyl group, enhancing the extent of intramolecular hydrogen bonding and reduces the possibility of intermolecular hydrogen bonding formed with an aqueous solvent. A decrease in potential hydrogen bonds may increase the permeability of the peptide across the intestinal mucosa.[2]
In a study, using the enzyme sensitivity of H-Trp-Ala-Gly-Gly-Asp-Ala-OH to cyclic esters, the investigators developed the N-terminal amino-and C-terminal carboxyl-3- (2 ′ -carboxyl-4 ′, 6 ′ -diphenyl) -3,3-dimethylpropionic acid 2. These cyclic precursors are sensitive to esterase metabolism (slow steps), open rings in the body, and release linear polypeptide drugs.[3]Both cyclic precursor drugs were quantitatively degraded to a linear hexapeptide in 37 °C of pH 7.4 buffer. In human plasma, hydrolysis of cyclic precursor drugs 1 and 2 was significantly faster than in buffer. The two cyclic peptide precursors were significantly more metabolic stable in a biological environment (such as human blood), and the permeability of the intestinal mucosa was 70-fold enhanced in an in vitro cell culture model.
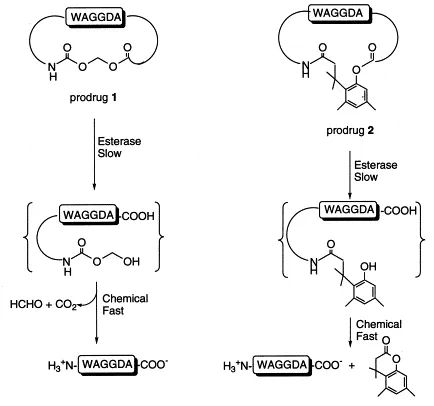
Figure 2. Mechanism of action of the cyclic peptide precursor drugs of H-Trp-Ala-Gly-Gly-Asp-Ala-OH. Photo credit: Adv.Drug Delivery Rev.[3]
Lopinavir (LVR, Lopinavir) is a protease inhibitor class of antiretroviral drug. Lopinavir and another protease inhibitor, ritonavir (lopinavir / Ritonavir) are used in a fixed-dose combination formula against HIV infection.
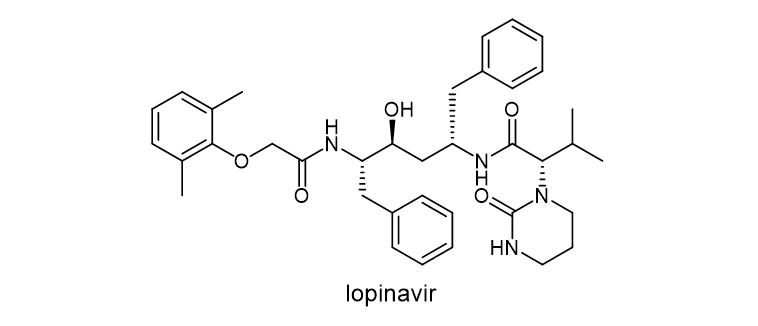
Figure 3. Chemical structure of lopinavir
Lopinavir is strongly metabolized by cytochrome P450 3A4 enzymes (CYP3A4) in vivo and prevented from entering cells by membrane efflux pumps such as P-gp and MRP 2. In order to avoid the first-pass metabolism and efflux of lopinavir, its two derivatives, valine-valine-lopinavir (VVL) and glycine-valine-lopinavir (GVL), were comparably tested as polypeptide precursors. The dipeptides Val-Val and Gly-Val were ligated to the side chain hydroxyl group of lopinavir, forming the VVL and GVL polypeptide precursor drugs, respectively.[4]

Figure 4. Lopinavir (LVR) polypeptide precursor drug VVL and GVL.
Enzymatic stability studies in Caco-2 cell homogenates show that the peptide precursor drug is first converted to an ester intermediate (amino acid precursor drug VL) and then finally to the parent drug LVR. Both VVL and GVL showed better solubility, metabolic, and intestinal permeability characteristics than the parent drug LVR. Since these precursor drugs are substrates for peptide transporter proteins, they can bypass efflux pumps to improve oral bioavailability. This advantage may also reduce the odds of developing resistance due to chronic exposure to LVR. Therefore, the effective delivery of polypeptide precursor drugs of LVR can improve the therapeutic effect, which has high clinical significance in HAART (highly effective antiretroviral therapy). A prodrug approach using peptide derivatives of LVR may be effective strategies to optimize the pharmacokinetic profile of LVR.
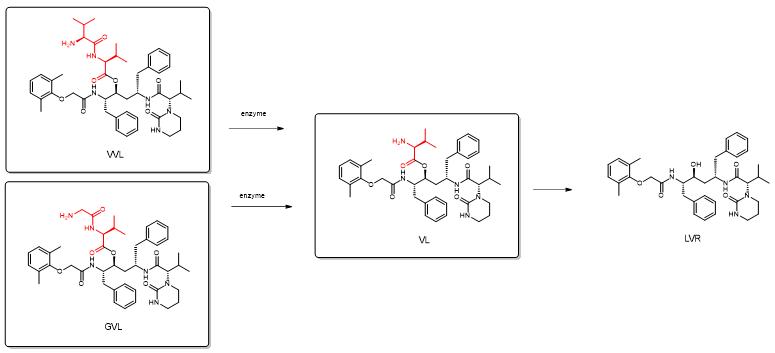
Figure 5. Reaction process of the polypeptide precursor drug VVL (LVR) with GVL.
sum up
In contrast to the permanent change in the structure of the polypeptide drug, the polypeptide precursor drug strategy focuses on temporarily changing the structure of the peptide molecule, so as to change the physical and chemical properties and metabolic performance of the molecule, thus increasing the permeability of the intestinal mucosa and improving the metabolic stability of the molecule. With the enthusiasm for the development and development of oral peptide drugs, the polypeptide precursor drug strategy will also usher in a new stage of development to promote the ultimate goal of oral peptide drugs.
reference material:
[1] A.H.Kahns, A.Buur, H.Bundgaard.Prodrugs of peptides.18. Synthesis and evaluation of various esters of desmopressin (dDAVP).Pharm.Res .1993, 10, 68-74.
[2] R.A.Conradi, A.R.Hilgers, N.F.H.Ho, P.S.Burton.The influence of peptide structure on transport across Caco-2 cells.II.Peptide bond modification which results in improved permeability.Pharm.Res., 1992, 9, 435-439.
[3] Giovanni M.Pauletti et al.Improvement of oral peptide bioavailability: Peptidomimetics and prodrug strategies.Adv.Drug Delivery Rev .1997, 27, 235-256.
[4] Sheetal Agarwal et al.Peptide prodrugs: Improved oral absorption of lopinavir, a HIV protease inhibitor.Int.J.Pharm.2008, 359, 7-14.
