

The FDA created an accelerated approval pathway (AA) in 1992 in response to the AIDS crisis, and subsequently used it for cancer treatment, and the EU established a conditional marketing license (CMA) in 2006, similar to the accelerated approval of the FDA.
China has launched a conditional approval (CA) pilot program since 2015, which means that new drugs can be approved earlier based on surrogate or intermediate clinical endpoints, similar to the FDA AA and EMA CMA programs.
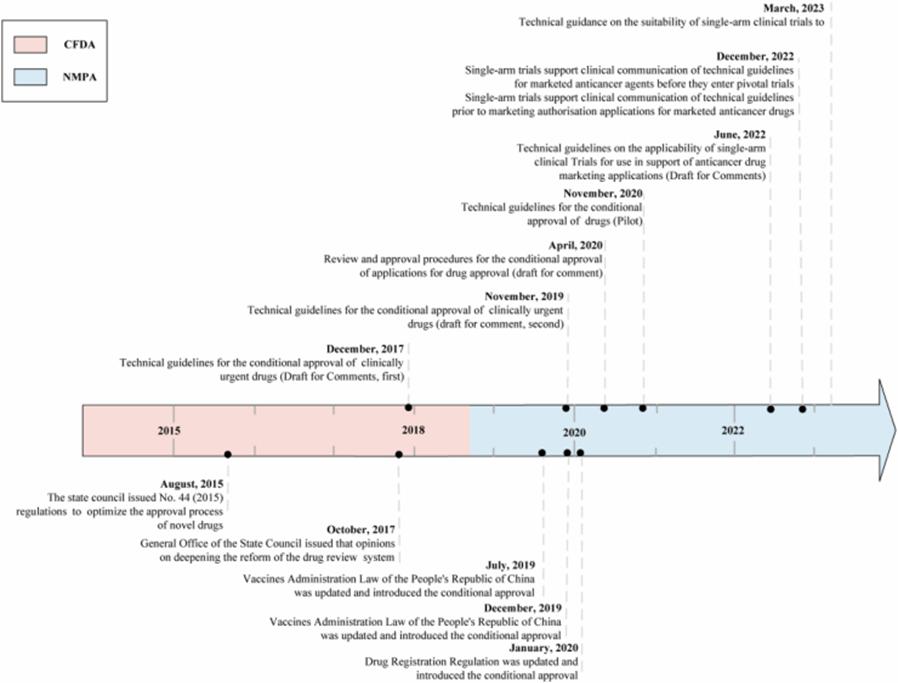
Figure 1: Scheduleline conditionally approved by China Drug Administration
The FDA requires AA drugs to complete confirmatory clinical trials within a specified time frame to determine whether to switch to routine approval or withdraw from the market, but nearly half of AA drugs show no clinical benefit, and AA currently appears to be the "death free gold" for some drugs. Due to the design limitations and / or its untimely completion of postmarketing studies, this uncertainty may persist for years after initial approval. See: the accelerated approval | nearly half of the FDA approved cancer drugs did not show clinical benefits
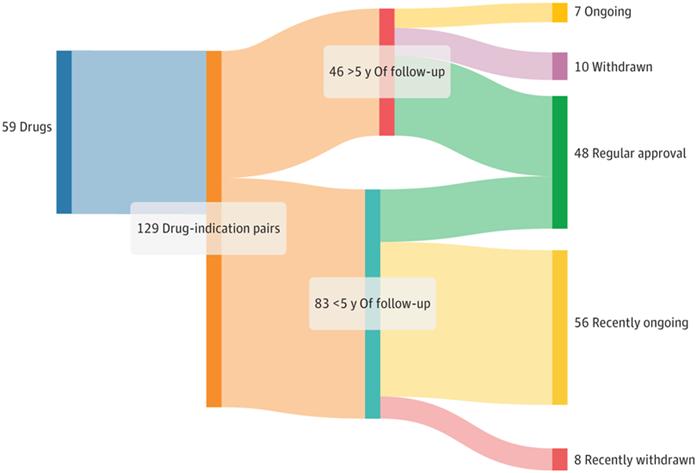
Figure 2: Regulatory results of accelerated approval of oncology drugs from 2013 to 2023
The FDA's AA is controversial: although the drug has reached its protocol-defined primary endpoint (mostly surrogate), the clinical benefits of the drug remain questionable. As mentioned above, AA became the "death free gold" for drugs, and the indications were retained in drug labeling and clinical practice guidelines for several years before being withdrawn.
As the application of CA in the cancer field gradually increases, China may also face these challenges. And 56 new anticancer drugs granted by NMPA to CA between 2015 and 2022, totaling 72 indications. Of these, 38 (53%) cancer indications were approved only in China, and 32 (44%) and 30 (42%) cancer indications were also approved by the FDA and EMA, respectively.
Of the FDA approved indications (32), 59% (19 / 32) and 41% (13 / 32) received AA and routine approval, respectively. EMA, approved 30 cancer indications, 40% (12 / 30) and 60% (18 / 30) received CMA and routine approval, respectively. The proportion of drugs in CMA was lower than FDA (40% versus 59%), which may be partly due to EMA, granting only the initial indication for CMA, while FDA and NMPA allow initial and complementary indications for AA and CA drugs.
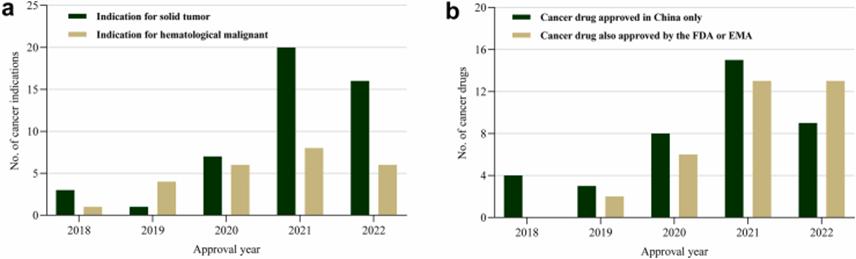
Figure 3: Conditional approval of cancer indications in China from 2018 to 2022
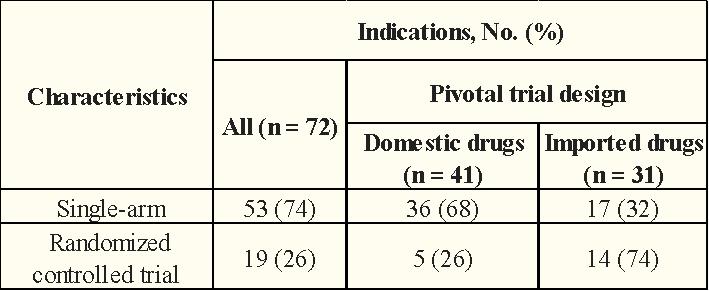
These pivotal clinical trials were mainly of a single-arm trial design (72%). Of the 72 pivotal clinical trials, RR was the primary endpoint (75%), followed by PFS (10%), OS (6%), and OS combined with PFS (4%).
Table 1: CA NMPA from 2015-2022 (Endpoint Design)
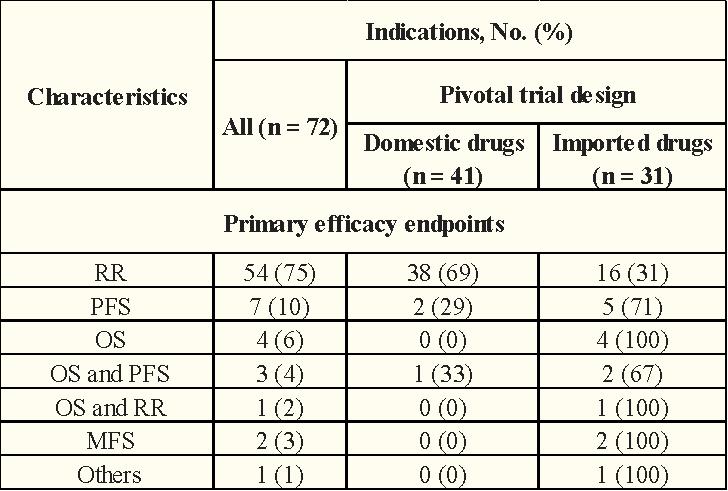
In terms of clinical trial design, the proportion of domestic anticancer drugs using a single-arm test was significantly higher than that of imported anticancer drugs (68% vs.32%)
Table 2: CA Anti-Cancer Drugs obtained with NMPA in 2015-2022 (Trial Design)
Validated clinical trials of anticancer drugs should be initiated before obtaining CA, and in addition, NMPA sets a rigorous timetable for the completion of confirmatory clinical trials of cancer drugs for CA. As more CA cancer drugs are approved for marketing, NMPA should further clarify the requirements for confirmatory clinical trial evidence, including the use of clinical endpoints and an RCT design whenever possible.
In contrast to US AA, China is still in the preliminary stage of the implementation of the CA program, and it is unknown whether these CA oncology drugs translate to clinical benefits in confirmatory clinical trials due to the lack of available data. To ensure that these anticancer drugs can be withdrawn or withdrawn from the market in the first place without clinical benefit, tracking the FDA's accelerated deregulation of drugs may provide some insight.
Most of the cancer drug indications accessing the FDA and subsequently withdrawn in the US had negative confirmatory clinical trial results, with the rest being incomplete confirmatory clinical trials or trials stopped due to safety concerns. All of these negative confirmatory clinical trials were designed as randomized controlled trials and included overall survival as a primary or secondary efficacy endpoint.
By the end of October last year, the FDA had 187 cancer indications available for AA, of which 65 are under validation, 96 are converted to conventional approval, and 26 indications have been withdrawn from AA since the launch of the program. Twenty-one of 26 cancer indications (81%) were based on a single arm study design; 17 (65%) were attributed to negative confirmatory clinical trial results, 7 (27%) to failure to complete confirmatory clinical trials, and 2 (8%) to safety concerns. The median duration from accelerated approval to final withdrawal was 4.1 years (IQR of 2.7-7.5).
Of the 26 cancer indications, 5 (19%) have been approved in China, 8 (31%) are in the new drug research phase, 3 (12%) have voluntarily terminated the new drug research phase, 1 (4%) has voluntarily withdrawn the new drug application phase, and 9 (35%) have not been approved in China (no new drug or new drug application information).
Table 3: Regulatory status of FDA withdrawn cancer indications and current regulatory status in China
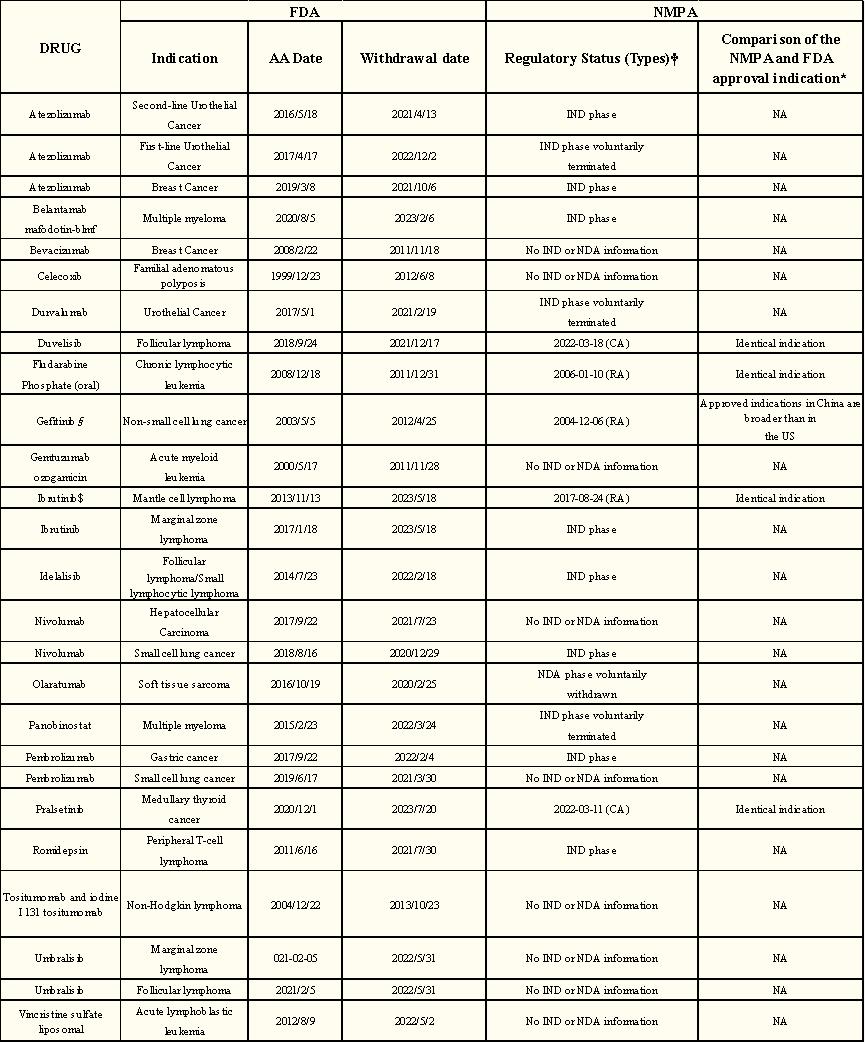
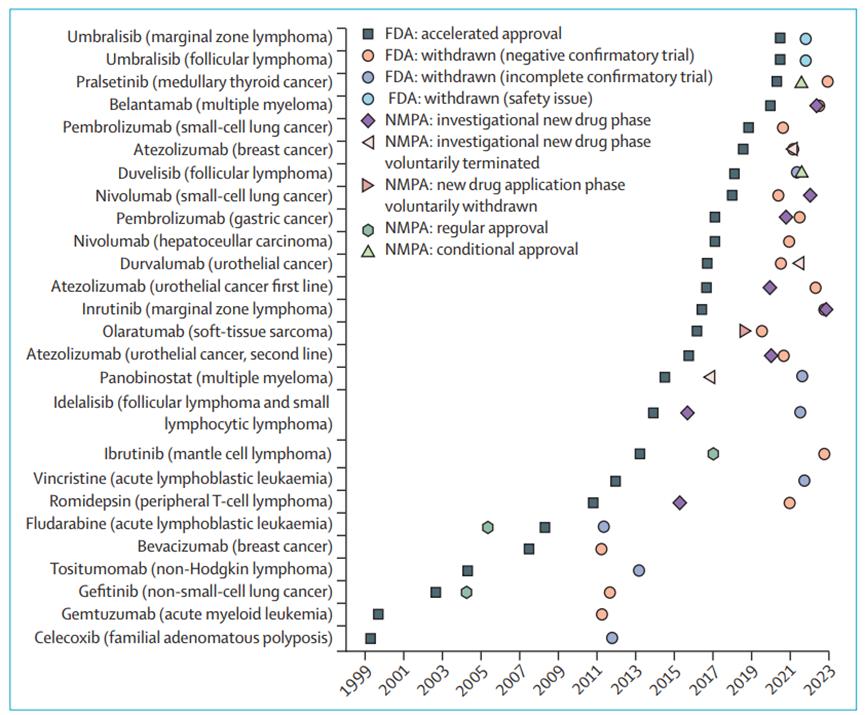
Figure 4: The FDA has withdrawn the regulatory status schedule for cancer indications
Currently, of the five approved indications available in China, only one was approved in China before accelerated FDA approval and the other four are approved in China after accelerated FDA approval. Of the five cancer indications, two received CA and three indications received routine approval (no conditional approval process was established at that time). Of these, 4 (80%) were approved later than those in the United States.
Of the 72 CA approved cancers with NMPA, 41 indications were produced by domestic manufacturers and 31 by imported indications. Of the 31 import indications, 30 were FDA approved and 28 were EMA approved, suggesting that FDA and EMA are consistent in benefit-risk assessment for most cancer drugs.
So the accelerated withdrawal of indications for cancer: what about the marketing licensing status in the EU?
As of April last year, the FDA approved accelerated approval of 23 cancer indications (18 different drugs) and was withdrawn, with 13 (57%) applying for similar marketing licenses to the EMA.
Of the 13 indications, 3 (23%) were denied marketing, 1 (8%) received conditional marketing, but was later withdrawn, and the remaining 9 (69%) received similar indications (8 standard and 1 conditional marketing).
The number of indications for withdrawing accelerated approval is lower than that determined by EU countries. This discrepancy may be due to a longer submission delay in approving new cancer drugs in China than in Europe, as most of the accelerated approval indications withdrawn from the US are still in the new drug research or new drug application phase in China or have not yet planned for marketing in China.
Table 4: Comparison of FDA approved cancer indications revocation accelerated approval with EMA approved related indications
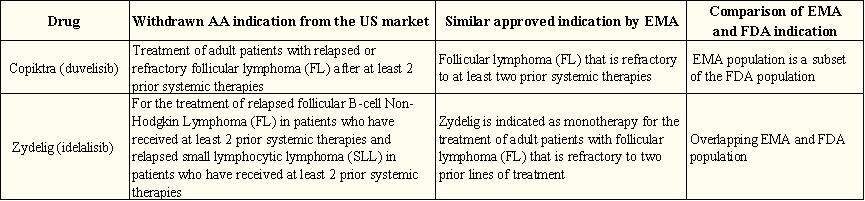
Take Duvelisib as an example of its accelerated approval / withdrawal in the US and the EU marketing license:
Duvelisib As a third-line treatment for follicular lymphoma, it received accelerated FDA approval in September 2018. To continue with the approval, the FDA requested a randomized phase 3 study with progression-free survival as the primary endpoint (the deadline for submission of the validation study report is November 2020). In May 2021, the Duvelisib obtained a standard marketing authorization from the EMA, based on the same evidence as the accelerated approval indication. This approval was based on a single-arm phase 2 study with an objective response rate for the primary endpoint.
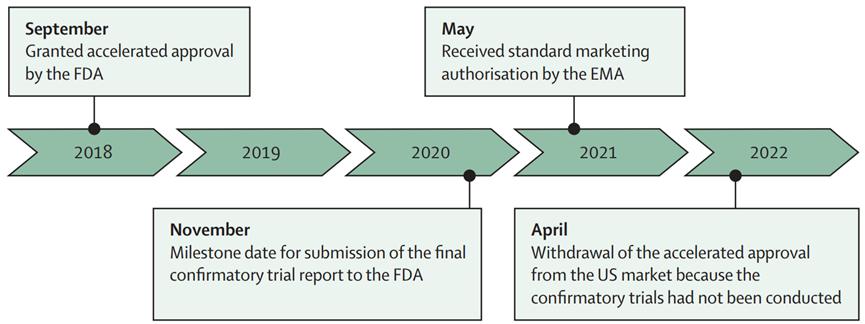
Figure 5: Approval schedule for accelerated approval / withdrawal and EU marketing licenses in the US (Duvelisib for example)
Of the 23 accelerated approved cancer indications that were withdrawn in the US between 1992 and April 2023,9 received marketing licenses for similar indications in the EU. In contrast, the EU revoked only 2 conditional marketing licenses for cancer indications in 2006 during the period of 20 April 2023, both of which were no longer approved in the United States.
EMA more often receives single-arm studies to confirm clinical benefits than FDA, while FDA more often requires randomised controlled trials. Both institutions rely mainly on alternative measures of patient benefit to meet their post-marketing obligations. After a median follow-up of 7 years, 40% of FDA and 61% of EMA post-marketing obligations after AA and CMA were delayed (the preliminary and limited nature of evidence submitted at initial approval brings uncertainty, which can be addressed by mandatory post-marketing studies).
These data show a discrepancy between the FDA, EMA, and NMPA approval decisions.
Institutional AA / CMA / CA, the benefit-risk balance of pathway indications may approach equal, leading to different drug fates; another reason that post-marketing studies may not adequately address the benefit-risk ratio uncertainty is the use of surrogate indicators such as PFS (the effectiveness of the widely used surrogate PFS in cancer is often still unproven) rather than patient-related outcomes, such as overall survival or quality of life;
And potentially differences between the times applied in the AA / CMA / CA pathway, this delay may lead to the withdrawal of some cancer indications for AA after completion of confirmatory clinical trials in the US that remain in confirmatory clinical trials in central Europe.
In fact, in China, not only to obtain FDA AA pathway tumor drugs, most tumor drugs NMPA is slower than FDA.
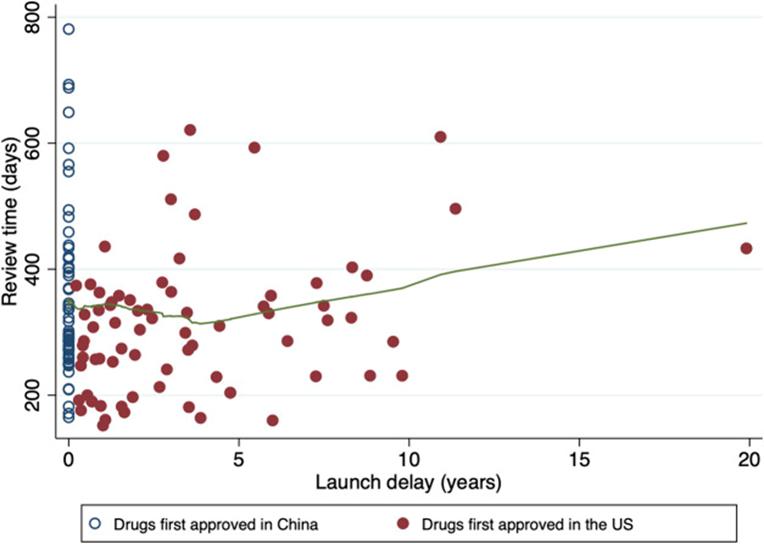
Figure 6: Time of drug review
Greater collaboration and data sharing among agencies will help reduce approval variation, especially for cancer drugs marketed through the AA pathway. Drug authorities need to do more to better strike a balance between reducing uncertainty and promoting the supply of promising new drugs.
reference material:
1.Luo X.Evidence of pre-approval clinical trial supporting the granted conditional approval for novel cancer drugs in China between 2015 and 2022. EClinicalMedicine.2023
2.Luo X.Current regulatory status in China of drugs withdrawn from the US FDA accelerated pathway: implications for regulation globally.Lancet Oncol.2024
3.Cramer A.Withdrawn accelerated approvals for cancer indications in the USA: what is the marketing authorisation status in the EU?Lancet Oncol.2023
4.Zhu X.Review time of oncology drugs and its underlying factors: an exploration in China.Front Pharmacol.2023
